简单的 GWAS 教程
假设有这样一个控制粒宽的 SNP,在一个 300 个品种的群体中,如果一个品种在这个位点是 C, 它的粒宽就相对较窄的,是 G 的话则相对较宽。
如果我们对这 300 个品种的粒宽性状进行测量,同时也测定它们在这个 SNP 上的基因型, 将 300 个品种分成 C 和 G 两组,用 T-test 检验粒宽上的差异,那么所得到的 P 值肯定会是一个很小的数字。
然而现实情况是,我们并不知道这样的控制粒宽的 SNP 在哪。我们所拥有的, 是在这 300 个品种的基因组上有 1000000 个 SNP 位点,通过测序,我们得到了 300 个品种的的基因型数据, 同时通过测量种子,得到了这 300 个品种的粒宽的表型统计数据。
如何寻找控制粒宽的 SNP 呢?一个最简单的方法就是像上面那样做 T 检验, 做上一百万次,SNP 的 P 值越小,它就越可能是我们想找的。
这就是一个最简化的全基因组关联分析(GWAS),一种通过将基因型数据与表型数据进行关联分析, 进而从中寻找与表型相关联的基因组区间,甚至定位相关基因的分析方法。当然,实际的 GWAS 的计算过程比这要复杂得多。
本文将利用 3K-RG 测序计划的基因型数据,以及公开的表型数据,做一个简单的 GWAS。
本文所用到的脚本已上传至配套的 Github 仓库
准备工作
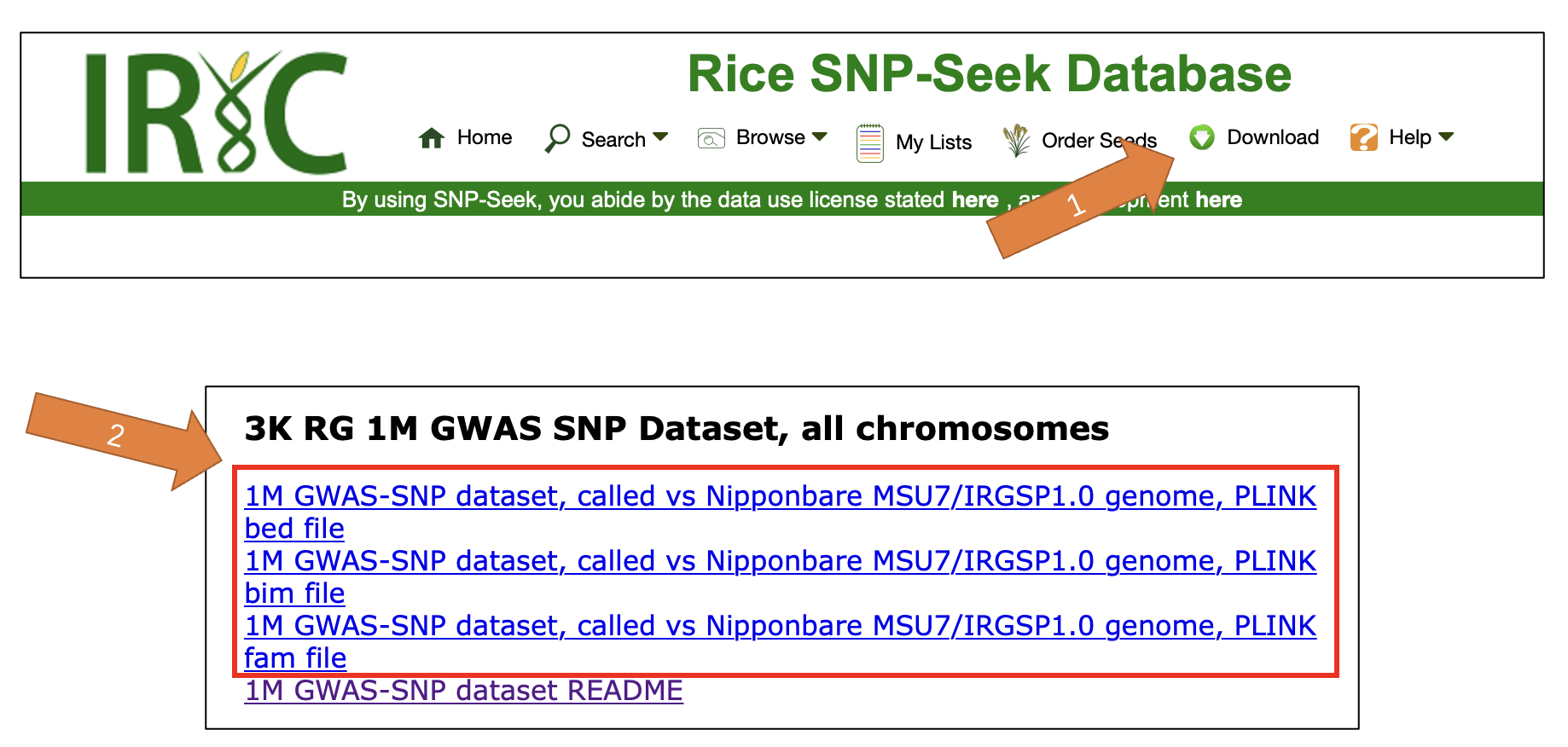
获取基因型数据
基因型数据可从国际水稻所的 SNP-Seek 数据库下载, 这里我们下载 “3K RG 1M GWAS SNP Dataset, all chromosomes” 下的数据, 这是 PLINK-BED 格式的数据,三个文件为一组数据,应始终放在一起。
- 二进制的 bed 文件存放着具体的每个品种的每个 SNP 位点是什么基因型这样的信息
- 纯文本的 fam 文件中存放着品种的编号和表型等信息(下载的 fam 文件里只有编号,表型数据是缺失值)
- 纯文本的 bim 文件中存放着 SNP 的物理位置等信息
有关 PLINK 格式的详细信息,可参考 PLINK 的文档。
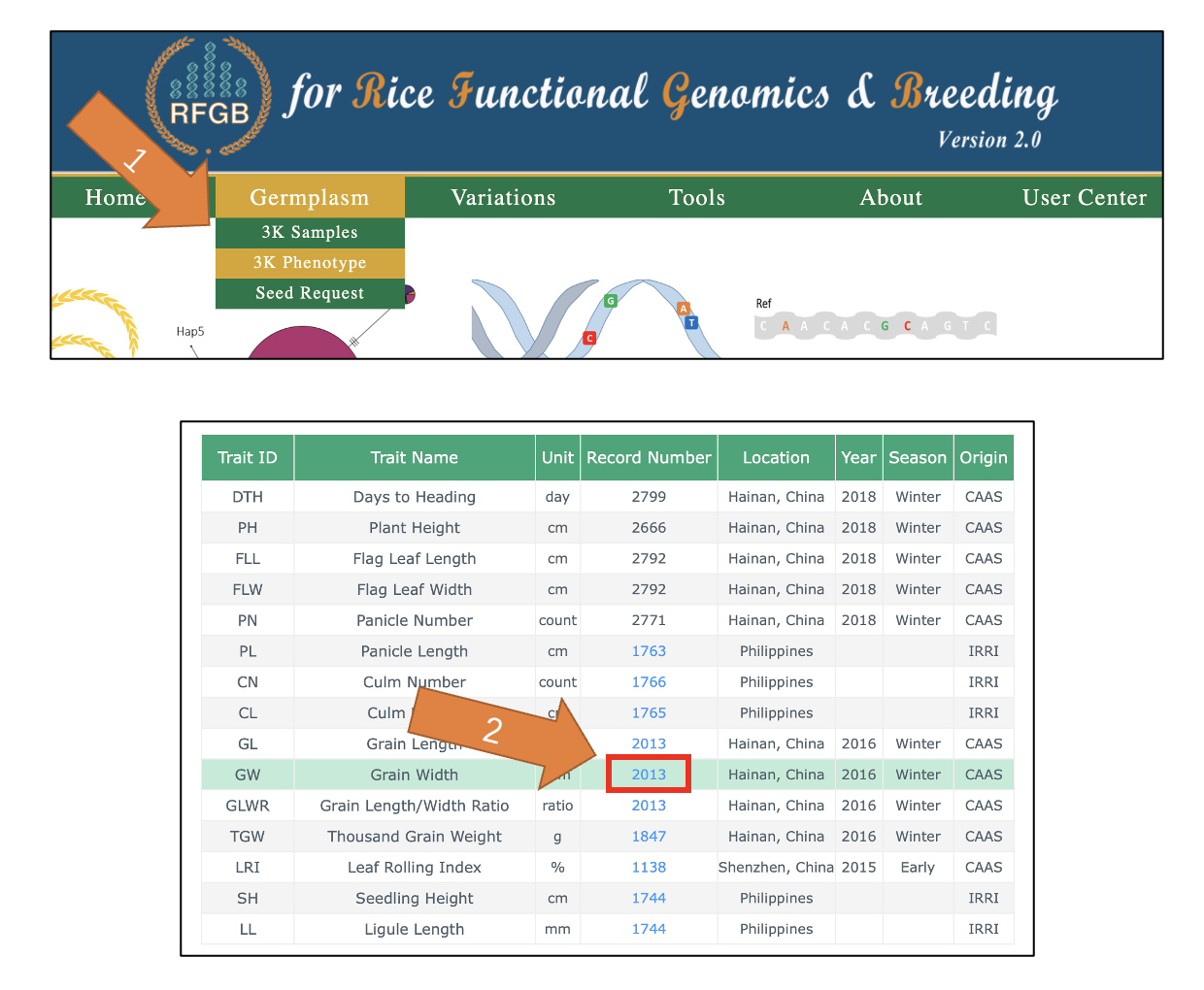
获取表型数据
表型数据可从 RFGB 数据库下载。 这里我们选择下载粒宽的数据。
安装软件
通过 conda 可快速安装需要的软件。首先将下面的 YAML 复制进一个文本文件中(比如 env.yaml)。
name: gwas
channels:
- bioconda
- conda-forge
dependencies:
- plink=1.90
- r-base=4.1
- r-tidyverse
- gemma
- r-qqman
然后通过下面的命令即可创建一个装好需要的软件的环境。
conda env create --file env.yaml
通过 conda activate gwas 即可激活环境。
建立项目文件夹
养成良好的习惯,创建一个项目文件夹,将下载下来的文件都放进来,形成如下所示的目录结构。
.
├── env.yaml
├── rawdata
│ ├── grain_width.zip
│ ├── pruned_v2.1.bed
│ ├── pruned_v2.1.bim
│ └── pruned_v2.1.fam
├── resources
├── results
└── scripts
分析流程
数据预处理
3K-RG 测序计划测了 3000+ 个品种,下载的粒宽表型数据也有 2000+ 个品种的数据, 作为一个教程,数据太大用个人电脑跑起来会很吃力,所以让我们随机地选出 300 个品种。
先用 R 语言脚本读入表型数据,并随机地挑出 300 个品种及其对应的表型数据,
这将生成一个格式与之前下载好的 fam 文件一样的文件:resources/selected.fam。
library(tidyverse)
grain_width <- read_tsv(
"rawdata/grain_width.zip",
col_names = c("ID", "Value")
)
set.seed(1)
selected_fam <- grain_width %>%
slice_sample(n = 300) %>%
transmute(FID = ID, IID = ID, Father = 0, Mother = 0, Sex = 0, Value)
write_delim(selected_fam, "resources/selected.fam", col_names = F, delim = " ")
然后,用 PLINK 以刚才生成的 fam 文件为名单,从下载下来的总的 PLINK-BED 格式数据中,
将 300 个品种的基因型信息提取出来,生成新的一组数据:resources/gwas.bed,
resources/gwas.bim 和 resources/gwas.fam。
plink \
--keep resources/selected.fam \
--bfile rawdata/pruned_v2.1 \
--make-bed \
--out resources/gwas
再用 R 语言脚本,用有表型数据的 resources/selected.fam 替换没表型数据的 resources/gwas.fam。
(在替换的过程中,也对 ID 的顺序进行了校对)
library(tidyverse)
col_names <- c("FID", "IID", "Father", "Mother", "Sex", "Value")
empty_fam <- read_delim("resources/gwas.fam", col_names = col_names, delim = " ")
selected_fam <- read_delim("resources/selected.fam", col_names = col_names, delim = " ")
sorted_values <- selected_fam$Value[match(empty_fam$IID, selected_fam$IID)]
sorted_fam <- empty_fam %>%
mutate(Value = sorted_values)
write_delim(sorted_fam, "resources/gwas.fam", delim = " ", col_names = F)
这样,用于 GWAS 的表型数据和基因型数据就准备好了。
PCA
理论上,并不是随便挑一些品种,测测表型就能够做 GWAS 的,如果品种间存在复杂的群体结构, GWAS 的结果很可能就有错误。我们可以通过绘制 PCA 图,来观察品种之间的关系。
首先用 PLINK,计算绘制 PCA 图所需的 EigenValue 和 EigenVector。
plink \
--bfile resources/gwas \
--pca \
--out resources/pca_matrix
这将生成两个文件: resources/pca_matrix.eigenval 和 resources/pca_matrix.eigenvec。
将生成的文件读入 R 中,用 ggplot 绘制 PCA 图(生成的文件为 results/pca.pdf)。
library(tidyverse)
eigenval <- scan("resources/pca_matrix.eigenval")
eigenvec <- read_delim(
"resources/pca_matrix.eigenvec",
col_names = c("FID", "IID", paste0("PC", 1:length(eigenval)))
)
pdf("results/pca.pdf")
ggplot(eigenvec) +
geom_point(aes(x = PC1, y = PC2)) +
labs(
title = "PCA Plot",
x = sprintf("PC1 Eigenvalue: %.2f", eigenval[1]),
y = sprintf("PC2 Eigenvalue: %.2f", eigenval[2]),
)
dev.off()
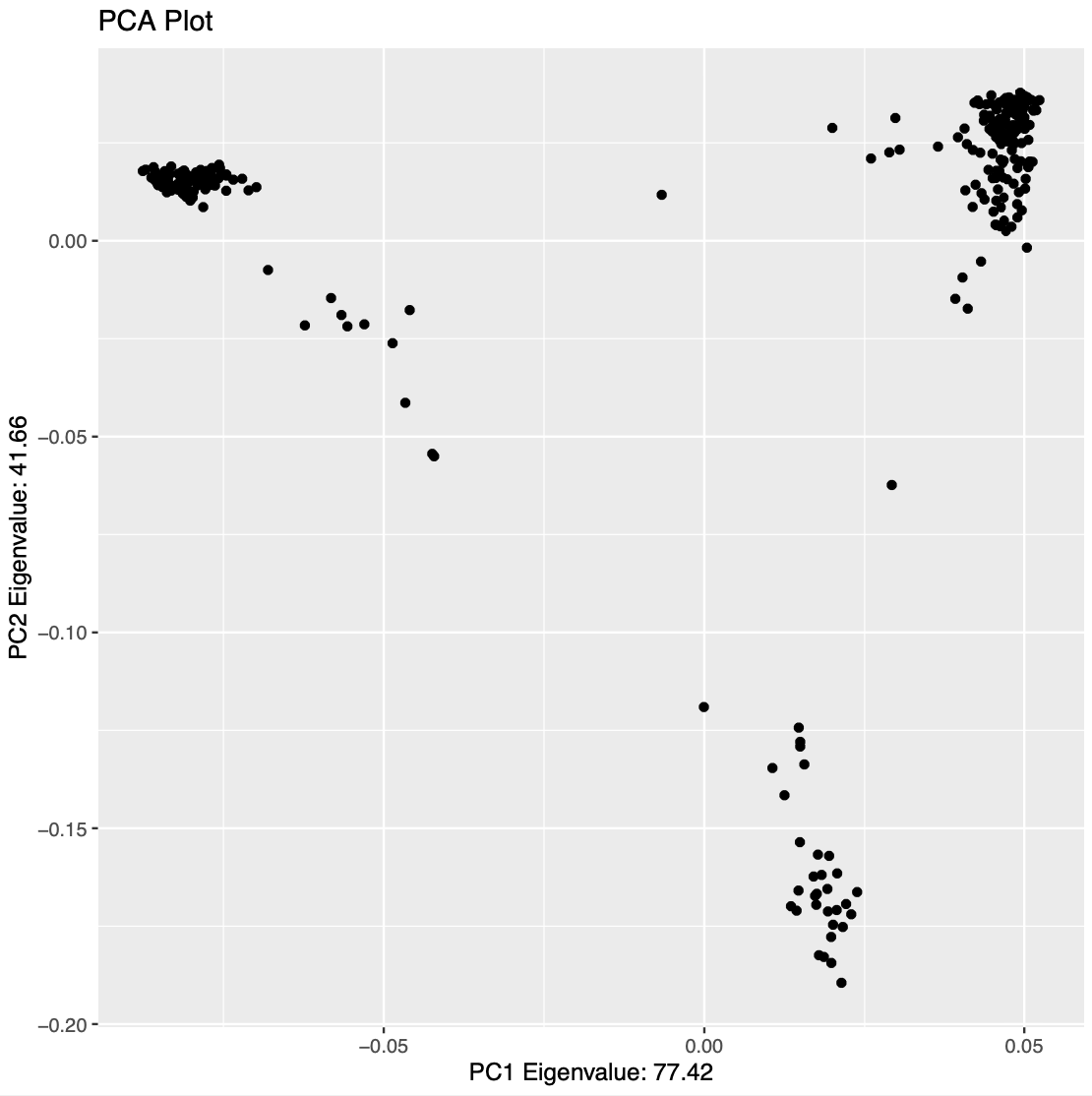
从图中可以看出,我们随机挑选的 300 个品种大致分成了三组,所以在后续的计算过程中, 应对群体结构进行校正。
关联分析
我们选用 GEMMA 用来进行计算每个 SNP 与表型关联的 P 值。
详细的软件文档可在命令行中键入
gemma -h或查阅在线文档。
首先,用 GEMMA 计算一个 300 个品种的亲缘矩阵,用以在后续的计算中矫正群体结构。
gemma \
-bfile resources/gwas \
-gk 1 \
-outdir resources
-o gemma_kinship
这将生成一个亲缘矩阵文件 resources/gemma_kinship.cXX.txt。
然后用 GEMMA 进行关联分析的计算。
gemma \
-bfile resources/gwas \
-k resources/gemma_kinship.cXX.txt \
-lmm 1 \
-outdir resources \
-o gemma_gwa
这将生成一个较大的纯文本表格文件:resources/gemma_gwa.assoc.txt,
内有 SNP 的相关信息,以及计算的 P 值。
Q-Q 图和曼哈顿图
虽然 GWAS 的大致原理就是对所有的 SNP 都进行关联分析,但实际上并不是一百万次 T 检验。
计算过程大概是这样的:先假设绝大多数 SNP 都与表型无实质关联, 并用我们所提供的基因型和表型数据对模型的参数进行估计, 使绝大多数 SNP 在这个模型里并没有与表型存在显著的关联,然后再把每个 SNP 都丢进这个模型里计算 P 值。 这样存在“超乎寻常”的关联的 SNP 就会有一个“超乎寻常”的低的 P 值。
Q-Q 图可用于展示模型拟合的优良程度,而在曼哈顿图中,SNP 的 P 值越小,纵坐标就会越高, 如果一个在一个较短的区域内有多个高度关联的 SNP,在曼哈顿图中就会形成一个关联的峰。
将之前计算所得的 resources/gemma_gwa.assoc.txt 读入 R,用 qqman 包即可绘制 Q-Q 图和曼哈顿图。
(生成 Q-Q 图文件:results/qq.pdf,生成的曼哈顿图文件:results/manhattan.pdf)
library(tidyverse)
library(qqman)
gwa_result <- read_tsv("resources/gemma_gwa.assoc.txt")
pdf("results/qq.pdf")
qq(gwa_result$p_wald)
dev.off()
pdf("results/manhattan.pdf")
manhattan(gwa_result, chr = "chr", bp = "ps", snp = "rs", p = "p_wald")
dev.off()
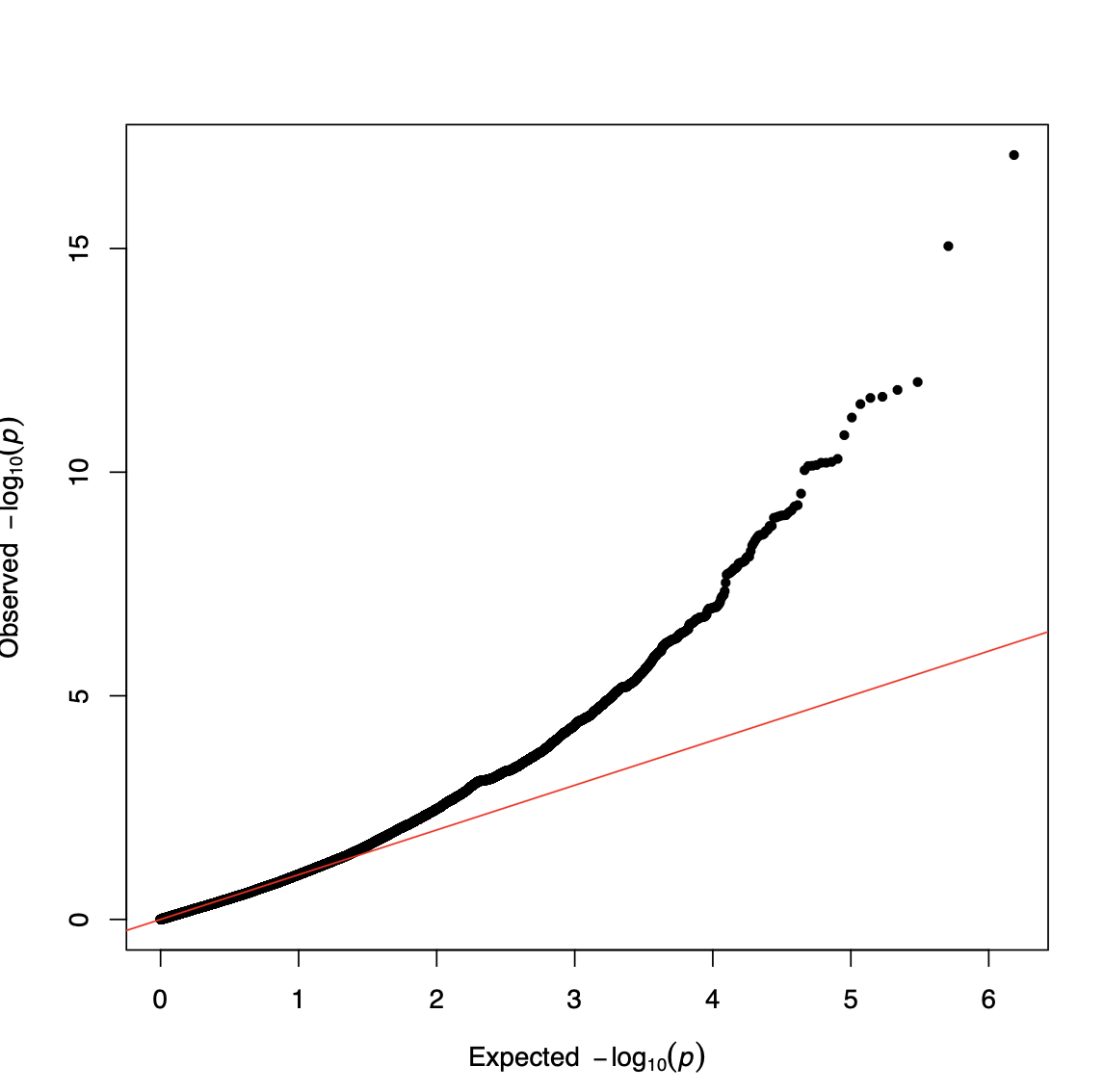
从 Q-Q 图中可以看出,大多数的 SNP 的 P 值都偏高,所以理论上关联分析的结果中将存在很多假阳性。
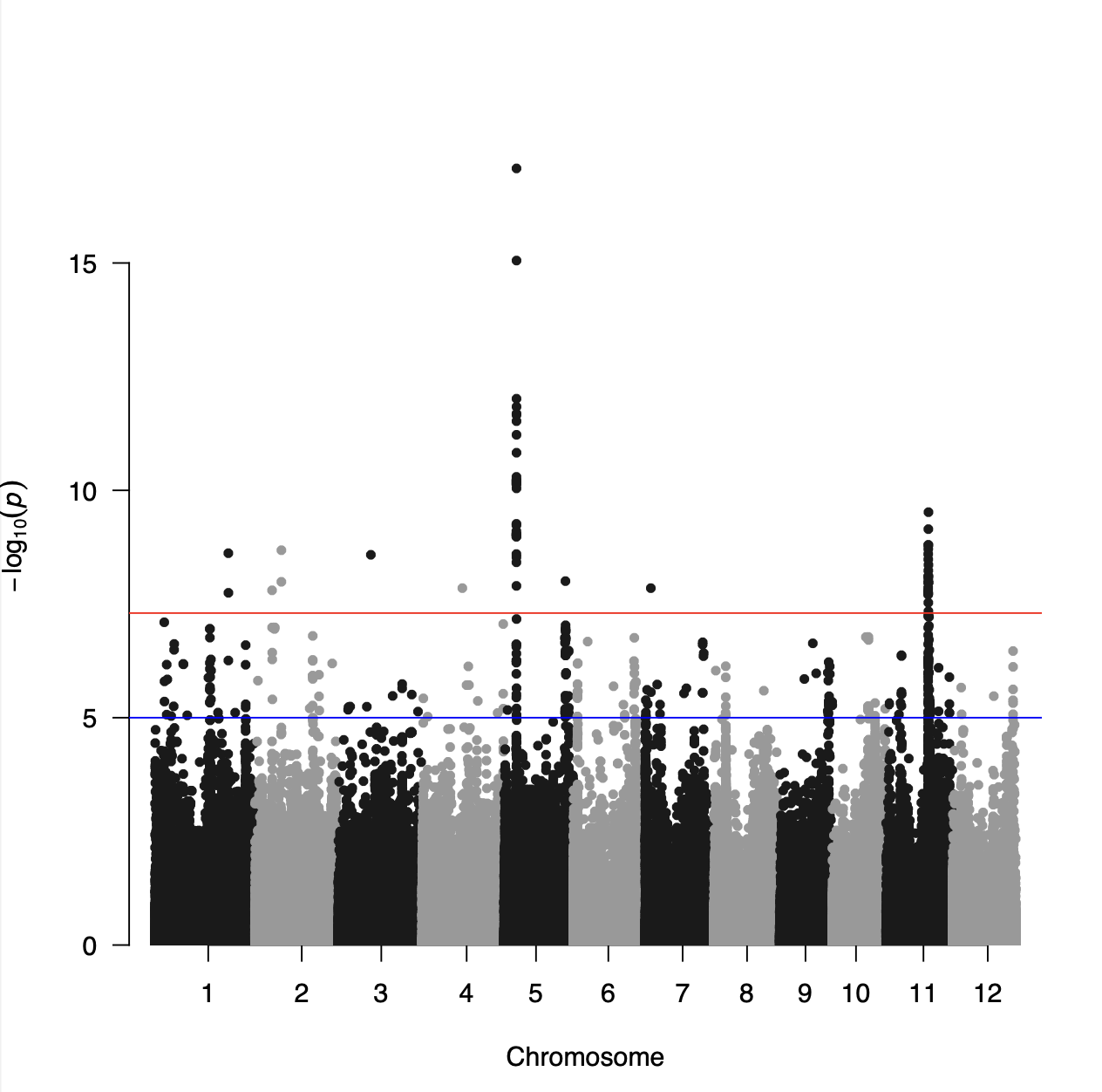
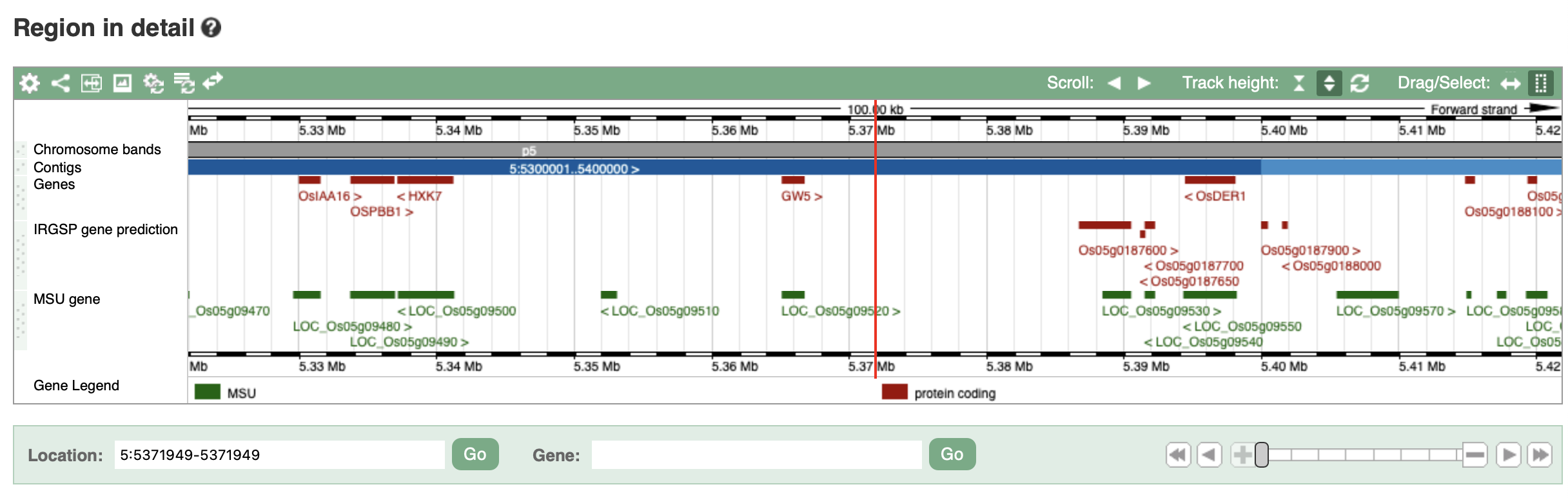
从曼哈顿图中可以看出,5 号染色体上存在一个非常强的关联信号, 我们可以用 R 脚本进一步查询 P 值最小的 SNP 的物理位置。
top10 <- function(chrom) {
gwa_result %>%
mutate(ps = as.character(ps)) %>%
filter(chr == chrom) %>%
arrange(p_wald) %>%
select(chr, ps, p_wald, allele0, allele1) %>%
slice_head(n = 10)
}
> top10(5)
# A tibble: 10 × 5
chr ps p_wald allele0 allele1
<dbl> <chr> <dbl> <chr> <chr>
1 5 5371949 8.11e-18 C G
2 5 5376161 8.84e-16 A G
3 5 5365256 9.67e-13 G A
4 5 5376243 1.45e-12 T A
5 5 5367026 2.06e-12 T A
6 5 5369058 2.19e-12 A G
7 5 5369111 3.01e-12 T A
8 5 5371214 6.01e-12 A G
9 5 5376245 1.49e-11 G A
10 5 5346621 5.05e-11 A G
通过查询 5:5371949 位置的 SNP 附近的基因,不难发现, 一个已知的控制粒宽的知名基因 —— GW5 就位于这个 GWAS 信号的附近。